Genomics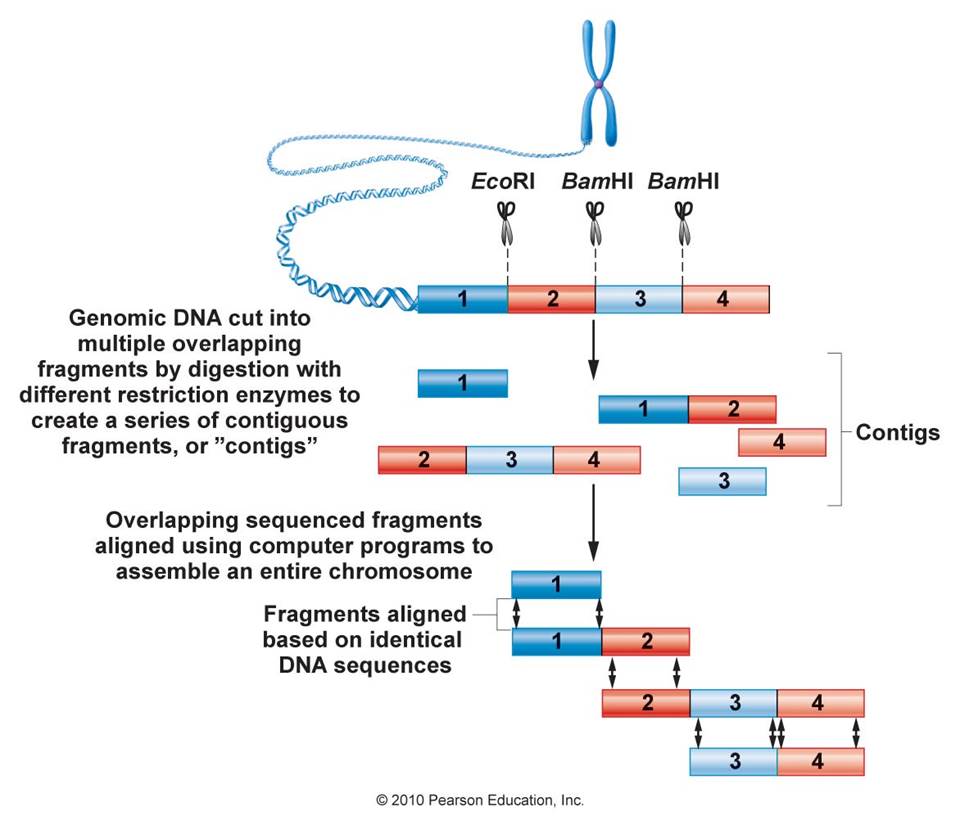
1) List three ways to make a DNA library.
2) Once you isolate the sequence of interest, you want to characterize it. Why is making a restriction map useful?
3) Suppose you conduct a restriction enzyme digest and produce the following results. Create a restriction site map.
| Lane | Fragment lengths | ||
|---|---|---|---|
| Control (no digest) | 6.0 kb | ||
| EcoRI | 2.5, 2.0, 1.5 | ||
| BamIII | 3.2, 2.8 | ||
| EcoRI and BamIII | 2.0, 1.5, 1.3, 1.2 | ||
| HindIII | 5.4, 0.6 | ||
| EcoRI and HindIII | 2.5, 2.0, 0.9, 0.6 | ||
| BamIII and HindIII | 3.2, 2.2, 0.6 | ||
| All 3 | 2.0, 1.3, 1.2, 0.9, 0.6 |
4) Describe the logic of adding dideoxy bases to the polymerization reactions to create fragments of differnt length. Why aren't ALL the adenine triphosphate precursors in the A lane dideoxy bases?
5) Suppose you sequence DNA using the Sanger method, and find the following banding pattern in your gel. What was the sequence?

5) Automated sequencers use the same logic, but they use single lane digests in capillary tubes, lasers, and dd-bases tagged with florescent markers. Exaplin how this works.
6) Copy the sequence below. Then, run a BLAST analysis (by pasting this sequence into the BLAST/FASTA window and BLASTing at the bottom of the page) to determine if this sequence is similar to anything at NCBI. What is it 100% similar too?
1 ggggcacccc tacccactgg ttagcccacg ccatcctgag gacccagctg cacccctacc
61 acagcacctc gggcctaggc tgggcggggg gctggggagg cagagctgcg aagaggggag
121 atgtggggtg gactcccttc cctcctcctc cccctctcca ttccaactcc caaattgggg
181 gccgggccag gcagctctga ttggctgggg cacgggcggc cggctccccc tctccgaggg
241 gcagggttcc tccctgctct ccatcaggac agtataaaag gggcccgggc cagtcgtcgg
301 agcagacggg agtttctcct cggggtcgga gcaggaggca cgcggagtgt gaggccacgc
361 atgagcggac gctaaccccc tccccagcca caaagagtct acatgtctag ggtctagaca
421 tgttcagctt tgtggacctc cggctcctgc tcctcttagc ggccaccgcc ctcctgacgc
481 acggccaaga ggaaggccaa gtcgagggcc aagacgaaga cagtaagtcc caaacttttg
541 ggagtgcaag gatactctat atcgcgcctt gcgcttggtc ccgggggccg cggcttaaaa
601 cgagacgtgg atgatccgga gactcgggaa tggaagggag atgatgaggg ctcttcctcg
661 gcgccctgag acaggaggga gctcaccctg gggcgaggtt ggggttgaac gcgccccggg
721 agcgggaggt gagggtggag cgccccgtga gttggtgcaa gagagaatcc cgagagcgca
781 accggggaag tggggatcag ggtgcagagt gaggaaagta cgtcgaagat gggatggggg
841 cgccgagcgg ggcatttgaa gcccaagatg tagaagcaat caggaaggcc gtgggatgat
901 tcataaggaa agattgccct ctctgcgggc tagagtgttg ctgggccgtg ggggtgctgg
961 gcagccgcgg gaagggggtg cggagcgtgg gcgggtggag gatgagaaac tttggcgcgg
1021 actcggcggg gcggggtcct tgcgccccct gctgaccgat gctgagcact gcgtctcccg
7) Now, go to the NCBI homepage and select "Sequence Analysis". Scoll down to "Open Reading Frame Finder". Paste the sequence into the large window and select 'ORF Find'.
8) There are six sequences presented, representing six different combinations of "open reading frames" - sequences stretching from a start codon to a stop, depending on whether the sequence is read "forward' (positive) or backward (negative). Click on the first colored box in the first diagram.