Modern Evolutionary Biology
I. Population Genetics
A. Overview
As a consequence of our modern understanding
of heredity and genetics, we have learned quite a bit about variation AND evolution,
and our model, a this point in the class is:
Sources of Variation
Agents of Change
MUTATION:
-New Genes:
Natural Selection
point mutation
Mutation (polyploidy can make new species)
RECOMBINATION:
- New Genes:
exon shuffling
-New Genotypes:
-crossing over
- independent assortment
 In
the early 20th century, at the same time that T. H. Morgan was studying mutations
and creating linkage maps, other biologists were considering the evolutionary
implications of this new knowledge regarding particulate, non-blending inheritance. They appreciated
that individuals do not evolve - evolution is a process that occurs at the population
level. For example, as a consequence of differential reproductive success among
individuals in a population, the range of phenotypes and their relative frequencies
in the population will change over time. Individuals are born, live, reproduce
(maybe) and die. As a result of passing on their genes at different frequencies,
the genetic structure of the population changes over time (evolution). Two biologists,
G. Hardy and W. Weinberg, constructed a model to explain how the genetic structure
of a population might change over time.
In
the early 20th century, at the same time that T. H. Morgan was studying mutations
and creating linkage maps, other biologists were considering the evolutionary
implications of this new knowledge regarding particulate, non-blending inheritance. They appreciated
that individuals do not evolve - evolution is a process that occurs at the population
level. For example, as a consequence of differential reproductive success among
individuals in a population, the range of phenotypes and their relative frequencies
in the population will change over time. Individuals are born, live, reproduce
(maybe) and die. As a result of passing on their genes at different frequencies,
the genetic structure of the population changes over time (evolution). Two biologists,
G. Hardy and W. Weinberg, constructed a model to explain how the genetic structure
of a population might change over time.
Their model begins by constructing
an 'equilibrium' model - a model of what the genetic structure would look like,
and how it would behave, if there was NO CHANGE over time. (We can liken this
to a "statistical null hypothesis of no effect", right?). Then, an actual
population is compared to this model, to see whether the population is evolving
or not.
B. The Genetic Structure of a Population
Our first step is to describe the
genetic structure of a population; we need to do this before we can model what
it would do over time. The genetic structure of a population is defined by the
gene array and the genotypic array. To understand what these are, some definitions
are necessary:
1. Definitions:
- Evolution: a change in the genetic structure of a population
- Population: a group of interbreeding organisms that share
a common gene pool; spatiotemporally and genetically defined
- Gene Pool: sum total of alleles held by individuals in a population
- Genetic structure: Gene array and Genotypic array
- Gene/Allele Frequency: % of alleles at a locus of a particular
type
- Gene Array: % of all alleles at a locus: must sum to 1.
- Genotypic Frequency: % of individuals with a particular genotype
- Genotypic Array: % of all genotypes for loci considered;
must = 1.
2. Basic
Computations - Determining
the Genotypic and Gene Arrays:
 The
easiest way to understand what these definitions represent is to work a problem
showing how they are computed.
The
easiest way to understand what these definitions represent is to work a problem
showing how they are computed.
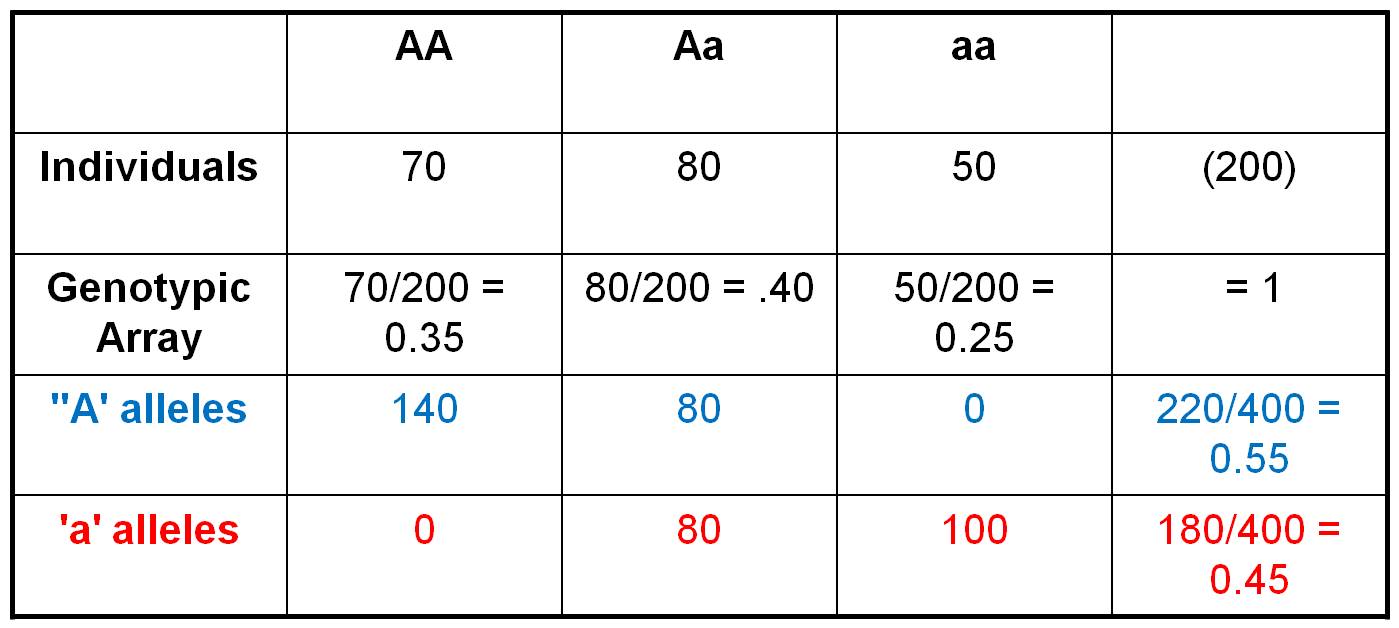
Consider the population shown to
the right, in which there are 70 AA individuals, 80 heterozygotes, and 50 aa
individuals. We can easily calculate the Genotypic Frequencies
by dividing each of these values by the total number of individuals in the population.
So, the Genotypic Frequency of AA = 70/200 = 0.35. If we account
for all individuals in the population (and haven't made any careless math errors),
then the three genotypic frequencies should sum to 1.0. The Genotypic
Array would list all three genotypic frequencies: f(AA) = 0.35,
f(Aa) = 0.40, f(aa) = 0.25. A Gene Frequency is the
% of all genes in a population of a given type. This can be calculated two ways.
First, let's do it the most obvious and direct way, by counting the alleles
carried by each individual. So, there are 70 AA individuals. Each carries 2
'A' alleles, so collectively they are 'carrying' 140 'A' alleles. The 80 heterozygotes
are each carrying 1 'A' allele. And of course, the 'aa' individuals aren't carrying
any 'A' alleles. So, in total, there are 220 'A' alleles in the population.
With 200 diploid individuals, there are a total of 400 alleles at this locus.
So, the gene frequency of the 'A' gene = f(A) = 220/400 = 0.55.
We can calculate the frequency of the 'a' alleles the same way. The 50 'aa'
individuals are carrying 2 'a' alleles each, for a total of 100 'a' alleles.
The 80 heterozygotes are each carrying an 'a' allele, and the 140 AA homozygotes
aren't carrying any 'a' alleles. So, in total, there are 180 'a' alleles out
of a total of 400, for a gene frequency f(a) = 180/400 = 0.45. The gene
array presents all the gene frequencies, as: f(A) = 0.55, f(a)
= 0.45.
There is a faster way to calculate
the gene frequencies in a population than adding up the genes contributed by
each genotype. Rather, you can use these handy formulae:
f(A) = f(AA) + f(Aa)/2
f(a) = f(aa) + f(Aa)/2
So, to calculate the frequency of
a gene in a population, you add the frequency of homozygotes for that allele
with 1/2 the frequency of heterozygotes. In our example, this would be:
f(A) = 0.35 + 0.4/2 = 0.35 + 0.2
= 0.55
f(a) = 0.25 + 0.4/2 = 0.25 + 0.2
= 0.45
Wow... that's a lot faster.
C. The Hardy-Weinberg
Equilibrium Model
1. Goal:
The goal of the "Hardy-Weinberg
Equilibrium Model" (HWE) is to describe what the genetic structure of the
population would be if NO evolutionary change occurs. Working independently,
Hardy and Weinberg realized
that the gene frequencies in a population will NOT change - will remain in EQUILIBRIUM
- if the following conditions are met:
- there is random mating
- no selection
- no mutation
- no migration
- and the population is infinitely
large.
And, they realized that a population
will reach an equilibrium in GENOTYPIC frequencies, too, after one generation
of meeting these expectations. And, for as long as these conditions are met,
a population will NOT EVOLVE. Let's see how they came by these conditions.
 2.
Example:
2.
Example:
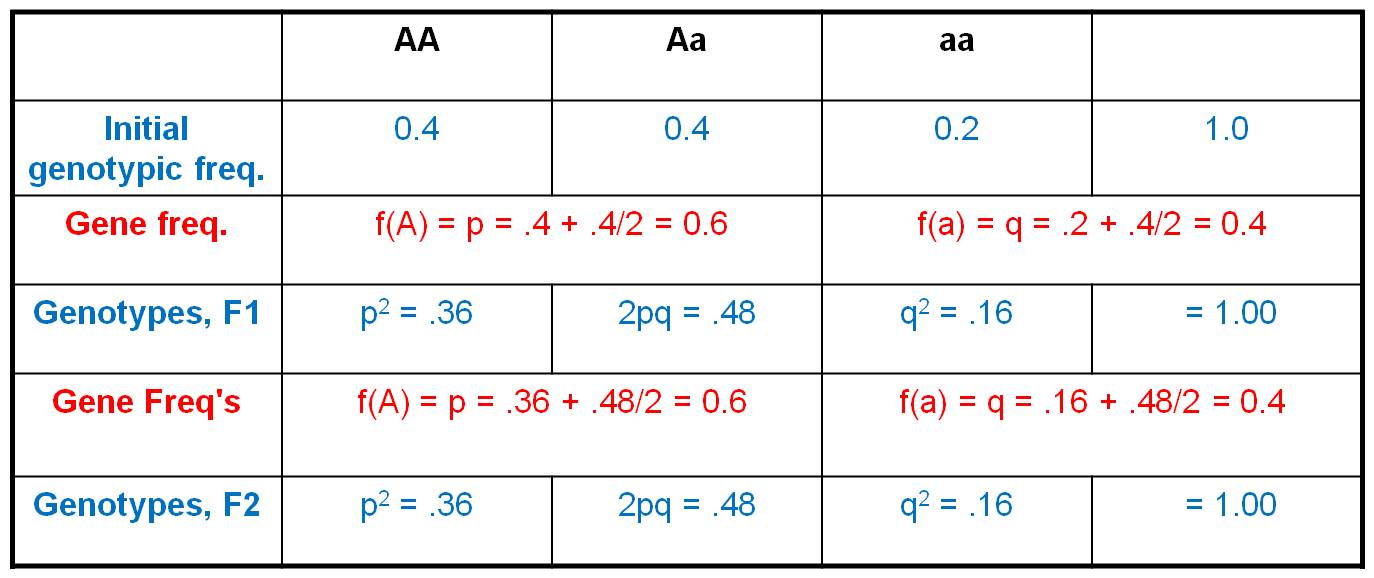
Consider an initial population, with
a genotypic array as shown. The gene frequencies are:
A = 0.4 + (0.4/2) = 0.6
a = 0.2 + (0.4/2) = 0.4
Now, consider this gene pool in which
60% of the alleles are 'A' and 40% of the alleles are 'a' (as defined by the
gene frequencies). The gene frequencies represent the frequencies of gametes
carrying these gens; so 60% of sperm are 'A', 40% are 'a', and likewsie for
eggs.
So, now we employ the HWE model.
IF the population mates at random, then we can use the product rule to determine
the probability of any two gametes coming together. The propability that and
'A' sperm fertilizes an 'A' egg = 0.6 x 0.6 = 0.36. And of course, this is the
only way to produce an 'AA' zygote. The frequency of 'AA' zygotes (the F1 offspring)
produced by this population should be 0.36. Likewise, the probability that an
'a' sperm fertilizes an 'a' egg = 0.4 x 0.4 = 0.16. And again, this is the only
way to make an 'aa' zygote, so the total frequency of 'aa' zygotes in the F1
will be 0.16. Now, there are two ways to make an 'Aa' zygote: an 'A' sperm can
fertilize an 'a' egg (probability = 0.6 x 0.4 = 0.24), and an 'a' sperm can
fertilize an 'A' egg (also with a probability of 0.4 x 0.6 = 0.24). So, the
total frequency of Aa zygotes in the F1 will be 2 x 0.24 = 0.48. If we generalize,
and let f(A) = p and f(a) = q, then the genotypic frequencies under HWE can
be calculated as: f(AA) = p2, f(Aa) = 2pq, and f(aa) = q2.
What is the genetic structure of
the population in the F1? Well, f(A) = f(AA) + f(Aa)/2 = 0.36 + 0.48/2 = 0.36
+ 0.24 = 0.6. And, f(a) = f(aa) + f(Aa)/2 = 0.16 + 0.48/2 = 0.4. So, the gene
frequencies did not change. And, if these organisms produce gametes at these
gene frequencies and mating is random, then F2 zygotes should be formed at the
frequencies of f(AA) = 0.36, f(Aa) = 0.48, and f(aa) = 0.16. Look familiar?
Indeed, after one generation of random mating, the population has reached an
EQUILIBRIUM - constant gene and genotypic frequencies over time.
Now, of course, these calculations
will only be true IF the population mates at random. AND, they will only be
true if there is no mutation. If 'A' alleles are mutating into 'a' alleles,
then the gene frequencies will not be 0.6 and 0.4, and calculations based on
these numbers will not be correct. So, we must assume NO MUTATION. Likewise,
we can't have any migration; we can't have 1000 AA individuals migrate into
our population, or that would change the gene frequencies, too; and our predictions
based on frequencies of 0.6 and 0.4 would be incorrect. So, we must assume NO
MIGRATION, too.
So, at this point we have zygotes
at the frequencies shown in the "Genotypes, F1" row. In order for
there to be no change in the genetic structure of the population, there must
be NO SELECTION. In other words, all genotypes must have the same probability
of survival and reproduction. Only then will they contribute gametes at frequencies
of p = 0.6 and q = 0.4. (If there were selection, and if AA individuals were
the only zygotes to survive to reproduce, for instance, then the gene frequencies
would change and our predictions based on frequencies of 0.6 and 0.4 would not
be correct).
And finally, this model will only
be explicitly true for populations that are infinitely large: because that is
the only time when we can be garaunteed that predictions based on random chance
will be exactly met. (Think about it this way... suppose I give you a coin that
is absolutely perfectly balanced. It IS PERFECTLY BALANCED. And suppose I ask
you, "how many times do you have to flip that coin to be ABSOLUTELY SURE
of producing a 50:50 ratio of heads to tails? Well, if you only flip it four
times, you know that, just by chance, you would often get 3 heads and a tail
or 3 tails and a head. And even if you flip it 10,000 times, you might get 5001
heads and 4999 tails, even though the coin is perfectly balanced. To be absolutely
garaunteed that the predictions of this probabilisitic model will be met
exactly, you must flip the coin an infinite number of times. Obviously, this
is a theoretical constraint because no population is infinitely large. But this
is a theoretical model of no change, so we can employ theoretical expectations.
The same is true of our 'expectation' of a perfectly balanced coin - this expectation
will only be met, for sure, in an infinitely large sample. Yet we continually
employ that expectation for a perfectly balanced coin, even in finite samples.
So, if you flip the coin 20 times, how many heads would you expect? Your answer
of 10 is a theoretical expectation.
So, that is why these assumptions
exist. It is only when ALL these are met that the genetic structure of a population
will not change. It is only when ALL these assumptions are met that a population
will NOT evolve. Wow. That should seem rather amazing. It is only when these
assumptions are ALL met that a population WON'T change. If any of these assumptions
is not met, a population's genetic structure WILL change... and that is evolution.
So, from this analysis, we should expect populations to evolve - it is only
under a rare combination of events (no, mutation, no selection, no migration,
random mating, and an infinitiely large population) that evolution WON'T happen.
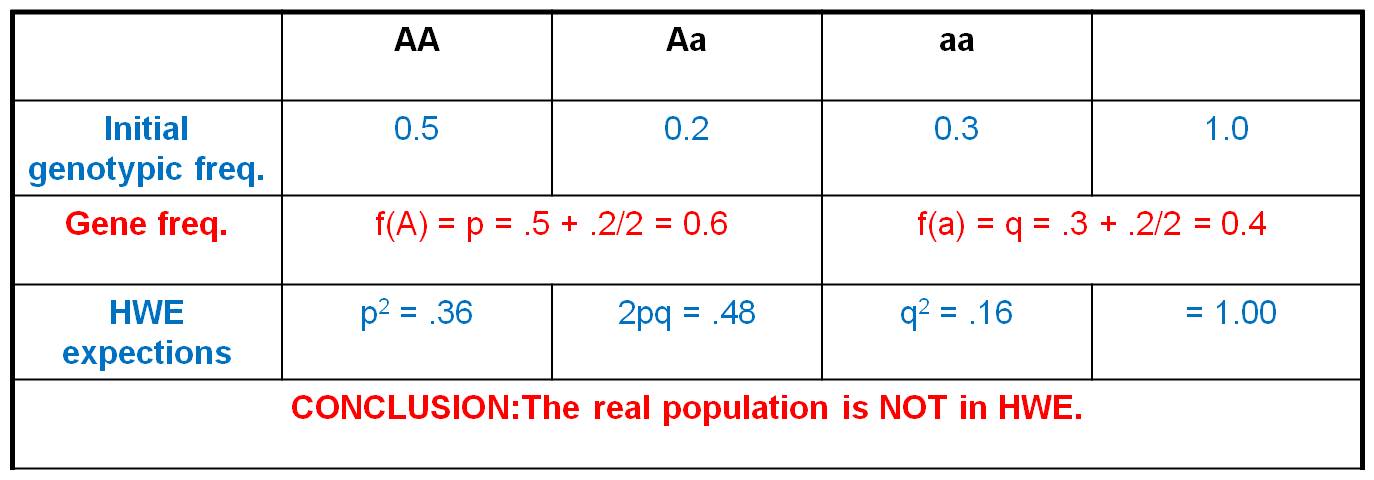
3. Utility
- If no real populations can explicitly
meet these assumptions, how can the model be useful? For instance, no
real population is infinitely large, so how can the model be useful? We
use it for COMPARISON. This model describes what the genotypic frequencies
should be IF the population was in equilibrium.
If the real genotypic frequencies are not close to these expectations, then
the population is not in HWE.... it is evolving. And if a population is not
in HWE, then the population must be violating one of the assumptions of the
HWE model. Think about that. The HWE is only 'true' if all the assumptions
are being met. If your real population differs from the model, then one
of the assumptions must not apply to your real population. This narrows
your focus on WHY the real populations isn't behaving randomly... and it might
identify WHY the population is evolving.... which is a biologically interesting
question.
 -
Again, the coin analogy applies. No REAL coin is probably exactly perfectly
balanced. But, if I give you a coin and ask you how balanced it is, you flip
it a few times and compare its behavior to WHAT YOU WOULD EXPECT FROM A PERFECT
COIN (50:50 RATIO). Even though a perfectly balanced coin may not exist, we
can use this theoretical model as a benchmark, to compare the behavior of real
coins. Many real coins act in a manner that is consistent enough with
the expectations from a perfectly balanced coin that we are willing to use them
AS IF they were perfectly balanced. The Hardy Weinberg Equilibrium
Model is the same... it is a theoretical model of no change against which we
can measure real populations.
-
Again, the coin analogy applies. No REAL coin is probably exactly perfectly
balanced. But, if I give you a coin and ask you how balanced it is, you flip
it a few times and compare its behavior to WHAT YOU WOULD EXPECT FROM A PERFECT
COIN (50:50 RATIO). Even though a perfectly balanced coin may not exist, we
can use this theoretical model as a benchmark, to compare the behavior of real
coins. Many real coins act in a manner that is consistent enough with
the expectations from a perfectly balanced coin that we are willing to use them
AS IF they were perfectly balanced. The Hardy Weinberg Equilibrium
Model is the same... it is a theoretical model of no change against which we
can measure real populations.
If HWE can be assumed, then the frequency
of recessive diseases can be assumed to equal q2, and the frequency of carriers
in the population can be estimated like this:
1) The frequency of hemachromatosis
worldwide is 1/450. If we assume that hemochromatosis is caused by a recessive
gene (q), and if we assume the population is in HWE with respect to this trait,
then q2 = 1/450 = 0.002. So, we take the square-root of both sides
to find q = 0.047. Well, if q = 0.047, and if p + q = 1, then p = 1 - 0.047
= 0.953.
2) If q = 0.047 and p = 0.953, then
the frequency of heterozygous carriers = 2pq = 0.09. So, we estimate that 9%
of the population are carriers.
Now, you might say, "but we
just determined that HWE would be unusual; so why would we assume it is
true for a given gene?" Well, a deleterious gene has already been
largely weeded out of a population, so selection against the few alleles that
are left is really weak. Indeed, this condition may not influence reproductive
success, anyway (NO SELECTION). In addition, we don't select mates based on
whether they have hemochromatosis (I bet you NEVER asked your date if they have
hemochromatosis, for example!!), so we can assume there is RANDOM MATING in
the population with respect to this trait. And although the human population
is not infinite, it is really big (~7 BILLION), so the effect of sampling error
is probably very small. Mutation is very rare, so the effects of mutation are
likely to be very small. And if we are making an estimate based on the whole
human population, then there can be no 'migrants' coming in from somewhere else
(Martians?). So, in some cases, we can reasonably assume a population might
be in HWE for a given gene. Of course, we could be wrong... and we would test
that prediction by sampling individuals in the population and determining the
frequency of heterozygotes genetically. But at least we would have a working
hypothesis.
As we've seen, equilibrium can only
occur if ALL of the assumptions are met. If they are not met, then the population
will evolve. We are now going to look at each assumption, and consider what
happens when each assumption is violated. This will show us how evolution can
occur by each of these different agents of evolutionary change.
D. Deviations
From HWE:
1. Mutation
Although large scale mutations like
polyploidy can cause instantaneous speciation, what we are talking about here
are substitution mutations that change one allele into another, or make a new
allele. Although such changes are very important
sources of new variation, they do not change the genetic structure of a population
very much at all, even when they occur: these mutations are rare, usually occurring
at a rate of 1 x 10-4 to 1 x 10-6.
Consider a population with:
f(A) = p = 0.6
f(a) = q = 0.4
Suppose 'a' mutates to 'A'
at a realistic rate of: µ = 1 x 10-5 . How will this rate of
mutation change gene frequencies? Not much: 'a' will decline by: qm = .4 x 0.00001
= 0.000004
'A' will increase by the same amount. So, the new gene frequencies will be:
p1 = p + µq = .600004, and q1 = q - µq = q(1-µ) = .399996.
So, mutation is a very important source of new alleles, but it doesn't change
the gene frequencies in a population very much.
2. Migration
Consider a resident population in
which p = 0.6 and q = 0.4. Suppose immigrants migrate into this population,
bringing A and a alleles into the population at these frequencies: p =0.8 and
q = 0.2. The effect of this influx will depend on the number of immigrants relative
to the number of residents. 100 immigrants may not change the genetic structure
of a population containing 1 million residents, but they could have a dramatic
effect on a population of 100 residents. We measure this relative effect by
quantifying the proportion of the total combined population that are immigrants.
So, in our example, suppose so many immigrants move in that they represent
10% of the new, combined population. We calculate new p as a weighted
average based on fraction of immigrants and residents:
So, p1 = (0.6)(0.9) + (0.8)(0.1)
= 0.54 + 0.08 = 0.62
residents contribute p at a rate of 0.6, and they represent 90% of the combined
population. Immigrants contribute p at a rate of 0.8, and they are 10%
of the population.
q1 = (0.4)(0.9) + (0.2)(0.1)
= 0.36 + 0.02 = 0.38, so we have done our math right because 0.62 + 0.38 = 1.0
There are two possible evolutionary
effects. First, migration will make two populations similar to one another;
particularly if the rate of immigration is high or the process is continuous
over time. Migration can also introduce new alleles into a population, but again
this effect will be correlated with the abundance of immigrants relative to
the number of residents.
 3.
Non-Random Mating:
3.
Non-Random Mating:
a. Positive Assortative Mating
There are many ways that non-random
mating can occur. We will look at a couple. The first example is called "positive
assortative mating". This is where mates 'sort' themselves with others
of the same genotype. This can be thought of as "like mates with like".
So, consider our old four o'clock plants with incomplete dominance. A population
might contain red, pink, and white flowers. Suppose the red flowers open in
morning, and are pollinated just by hummingbirds (that prefer red flowers).
Suppose the white flowers open at night, and are pollinated by moths. And suppose
the pink flowers open in the afternoon, and are pollinated by bees and butterflies.
In this case, "like mates with like" for flower color (and time of
opening). So, a plant with red flowers will only mate with another plant having
the same genotype for (red) flower color. Now, it is IMPORTANT to realize that
plants are only positively assorting for flower color and opening time in this
case. One red flowering plant may be tall while the other is short; one may
have hairy leaves while the other has smooth leaves. Indeed, the plants may
be mating at random with respect to all other traits.
When AA individuals mate only with
each other, all their offspring will be AA, as well. So, if 20% of the population
is AA (intial genotypic frequency = 0.2), and if there is no difference in reproductive
success (because we are only violating the assumption of random mating so there
is no selection), then these parents will make 20% of the offspring and they
will all be AA. The same goes for aa individuals only mating with other aa individuals
- all their offspring are aa. However, when Aa heterozygotes only mate with
one another, they produce AA, Aa, and aa offspring in a 1/4:1/2:1/4 ratio. If
60% of the population is heterozygous, then they will make 60% of the offspring...
but these offspring won't all be heterozygous; only 1/2 - or 30% will be heterozygous.
15% will be AA and 15% will be aa. So, the total frequency of AA offspring in
the F1 will be 35%; 20% had AA parents and 15% had Aa parents.
As a consequence of positive assortative
mating, the frequency of heterozygotes will decline and the frequency of homozygotes
will increase. Curiously, the gene frequencies won't change, so in the F1, f(A)
= .35 + 0.30/2 = 0.5... just as it was in the orginal population (f(A) = 0.2
+ 0.6/2 = 0.5). The genes are just being 'dealt' to offspring in a non-random
manner, affecting the genotypic frequencies at this locus.
So, suppose we observed the F1 population
in nature, and wanted to know if it was in HWE. We would calculate the gene
frequencies (A = 0.5, a = 0.5), and then estimate what the frequencies of the
genotypes would be IF the population was in HWE: p2 = 0.25, 2pq =
0.5, q2 = 0.25. We would compare our real population's genotypic
array with this HWE expectation, and see that they are not the same. And we
could see one thing more... we would see that the ACTUAL OBSERVED frequency
of heterozygotes (0.30) is LESS THAN the expected frequency of heterozygotes
under the HWE hypothesis (0.5). And we would see that the observed frequency
of both homozygotes is greater than expected. Knowing that positive assortative
mating can cause this pattern, we would have a working hypothesis regarding
the agent of evolution at work in this population.
b. Inbreeding: Mating with
a Relative
Inbreeding is mating with a relative.
It is similar to positive assortative mating, except that the two mates are
not just similar at one locus, but they are probably similar at MANY loci because
they are related and got their genes from the same ancestors. Siblings share,
on average, half their genes. Matings between siblings, then, will tend to reduce
heterozygosity at MANY loci, not just one.
The most extreme example is "obligate
self-fertilization". This is where a hermaphrodite ONLY mates with themselves.
This is not asexual reproduction - they produce gametes by meiosis and get all
the benefits of producing variable gametes that occurs in sexual reproduction;
but they only fertilize their own gametes. This has a profound effect on the
genetic structure of the population. Think about it: when an organism mates
with themselves, they are mating with an organism that has the SAME genotype
at EVERY locus. So, there will be a decrease in heterozygosity across the entire
genome, with a 50% reduction in heterozygosity each generation. This is the
most rapid loss possible. Siblings are only related, on average, by 50%, so
the loss of heterozygosity will only occur 1/2 as fast.... but it will still
occur at all loci across the genome.
Inbreeding often reduces reproductive
success, because there is an increase in homozygosity - and this means that
deleterious recessives are going to be expressed more frequently and exert their
negative effects on the offspring. A deleterious allele may be rare in a population,
but inbreeding will increase the probability that it occurs in the homozygous
condition and is expressed. Because inbreeding can reduce the survivorship of
offspring and thus reduce reproductive success of the parent, it is often selected
against. Selection favors different strategies that reduce the likelihood of
inbreeding, like "self-incompatibility" in some plants, like lions who push
male cubs out of a pride when they mature (thus they don't breed with their
sisters), or like humans who have a variety of cultural taboos against breeding
with relatives. However, inbreeding is also a mechanism for purging deleterious
alleles from the population. If a population can get through the first
few generations in which homozygote recessive are produced and selected against,
the net effect will be to eliminate these deleterious alleles from the population.
That can be a good thing in the long run.
Things to Know:
1.
What are the five assumptions of the Hardy-Weinberg Equilibrium Model?
Study Questions:
1.
Consider the following population:
| |
AA |
Aa |
aa |
| Number
of Individuals |
60 |
20 |
20 |
- -
calculate the genotypic frequencies.
- -
calculate the gene frequencies
- -
calculate the HARDY WEINBERG EQUILIBRIUM frequencies.
2.
If the HWE model does not describe any real population, how can it be useful?
Study Questions:
3.
Consider a population with p = 0.8 and q = 0.2. If the mutation rate of A-->
a = 4.0 x 10-6, what will the new gene frequencies be in the next
generation?
4.
Consider a population, p = 0.8 and q = 0.2. If migrants enter this population
with p = 0.1 and q = 0.9, such that immigrants comprise 15% of the total population,
what will the new gene frequencies be?
5. If the
population below undergoes positive assortative mating, what will the genotype
frequencies be in the next generation?