
This is the most dramatic change possible; an addition of an entire SET of chromosomes, changing a haploid gamete to a diploid gamete, or changing a diploid cell into a tetraploid cell.
1. Mechanism #1 - Complete failure of Meiosis
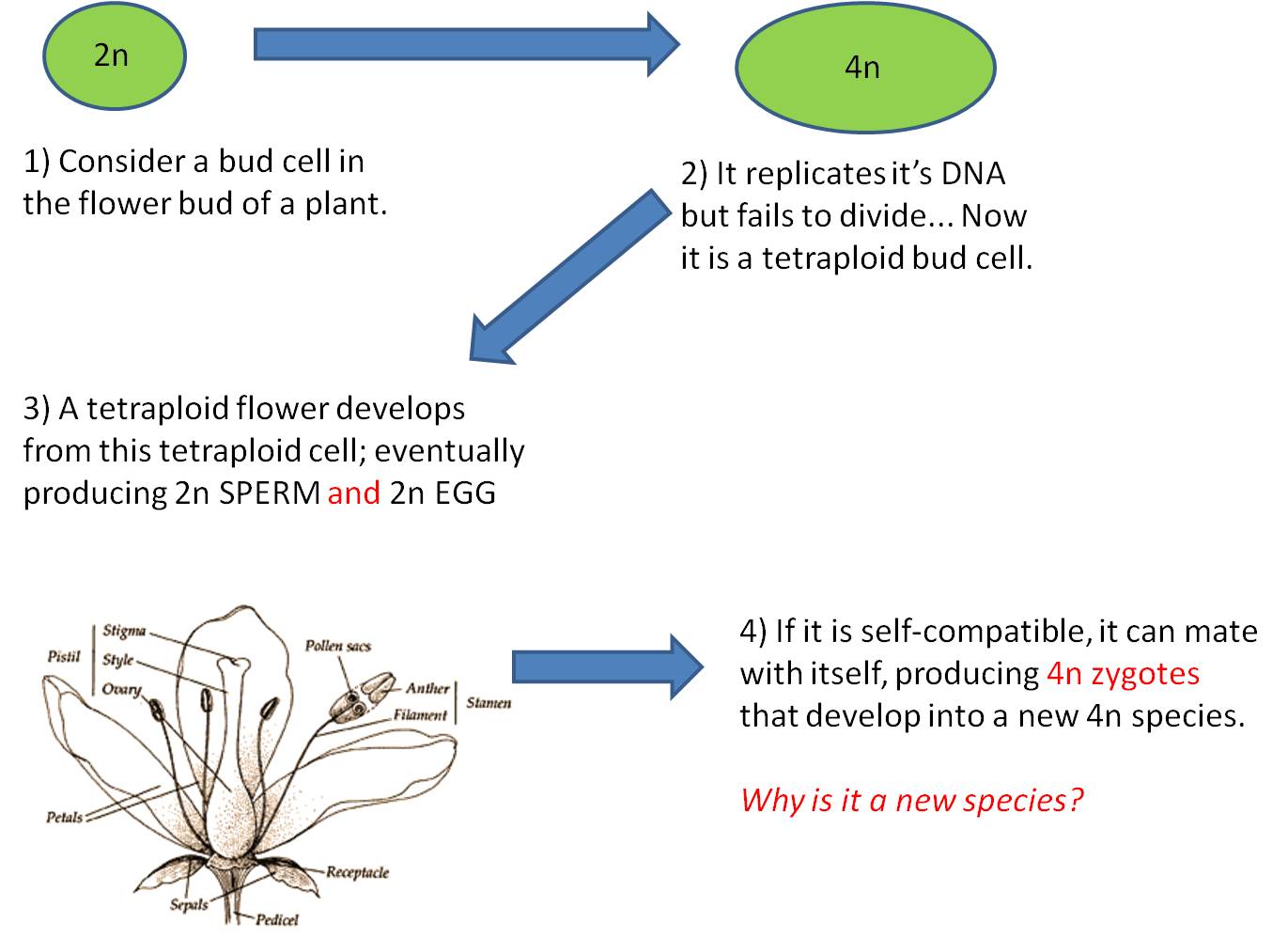 2.
Mechanism #2 - Failure of Mitosis
2.
Mechanism #2 - Failure of Mitosis
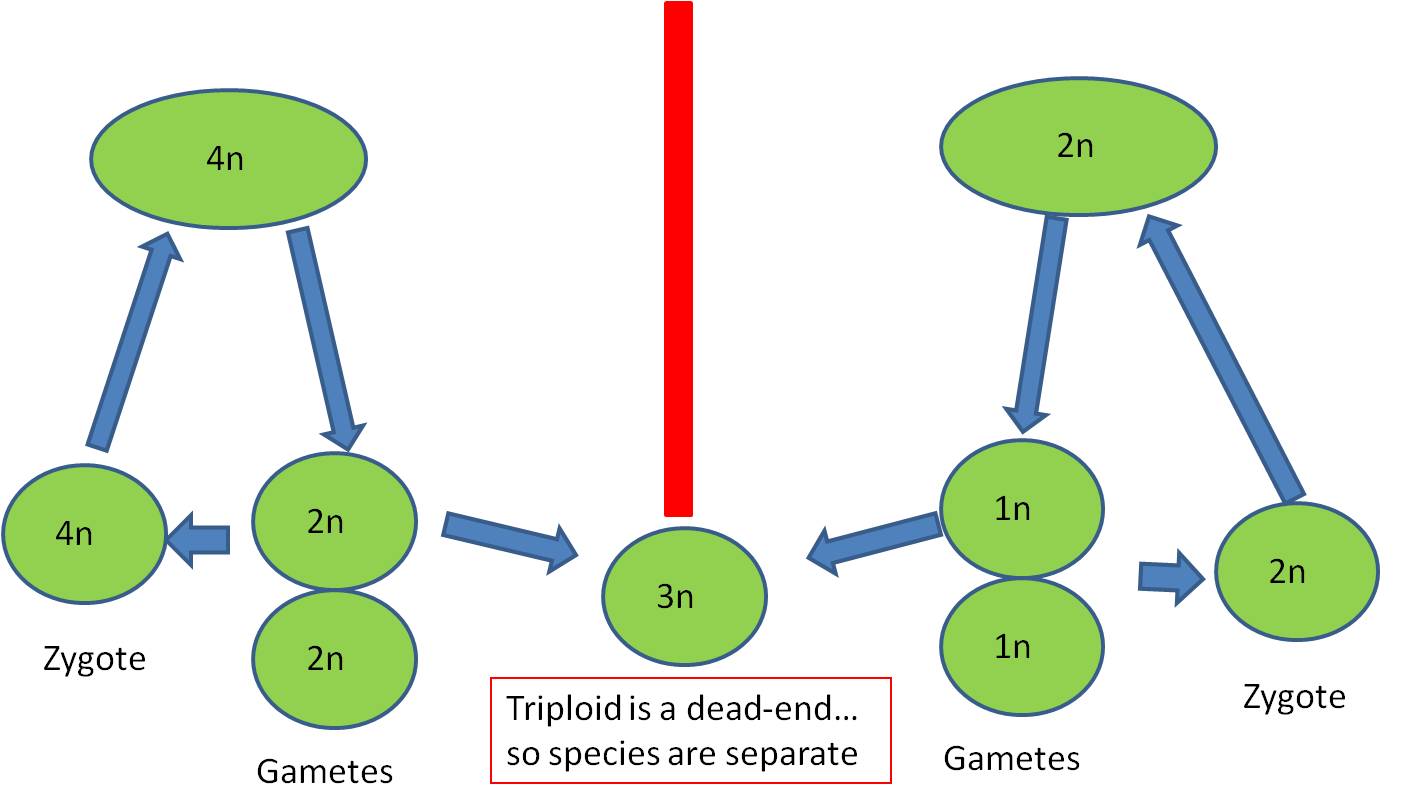
Here, however, there are 2n male AND 2n female gametes produced. If the plant is self-fertile, these sperm can fertilize these eggs and a 4n zygote will be produced. This is TETRAPLOIDY. - Tetraploid offspring may not survive; there may still be enough of a disruption in the protein concentrations to screw-up development. But, if they can survive, then they WILL be able to make gametes by normal meiosis. Their 4n cells CAN divide evenly, producing 2n gametes.
- This lineage could reproduce itself, and would be reproductively isolated from its originating species because IF their 2n gametes fertilized the haploid gametes of the originating species, a 3n triploid would be produced, which is typically a reproductive dead-end for reasons described, above.
 3.
Frequency of Polyploidy
3.
Frequency of Polyploidy
Polyploidy (>2n) is VERY rare in birds and mammals. The only known polyploid mammal is a small tetraploid mouse that lives in the Andes - it was discovered in 2002. No birds are known to be triploids. However, it is more frequent (but still rare) in fish, amphibians, and reptiles, where there are parthenogenetic 3n sister species of normal 2n species. However, on the whole, polyploidy is rare in dioecious species (where individuals are EITHER male or female). This is probably because the most likely polyploid product in these species is a triploid, which usually has the sterility problems mentioned earlier.
In hermaphroditic species (and particularly self-compatible plants), polyploidy is FAR more common. This is probably because tetraploidy is far more likely, and the tetraploid will be fertile. In fact, about 30-50% of all flowering plant species are polyploid!!!. That means that this mechanism has been a pretty important source of new species. Some genera have species with sequential increases in ploidy. For instance three different species of goldenrods have 14, 28, and 56 chromosomes, respectively. So, a reasonable hypothesis is that the second and third species were formed through polyploid events (failure in mitosis) as described above - doubling their ploidy levels and making new species.
 This
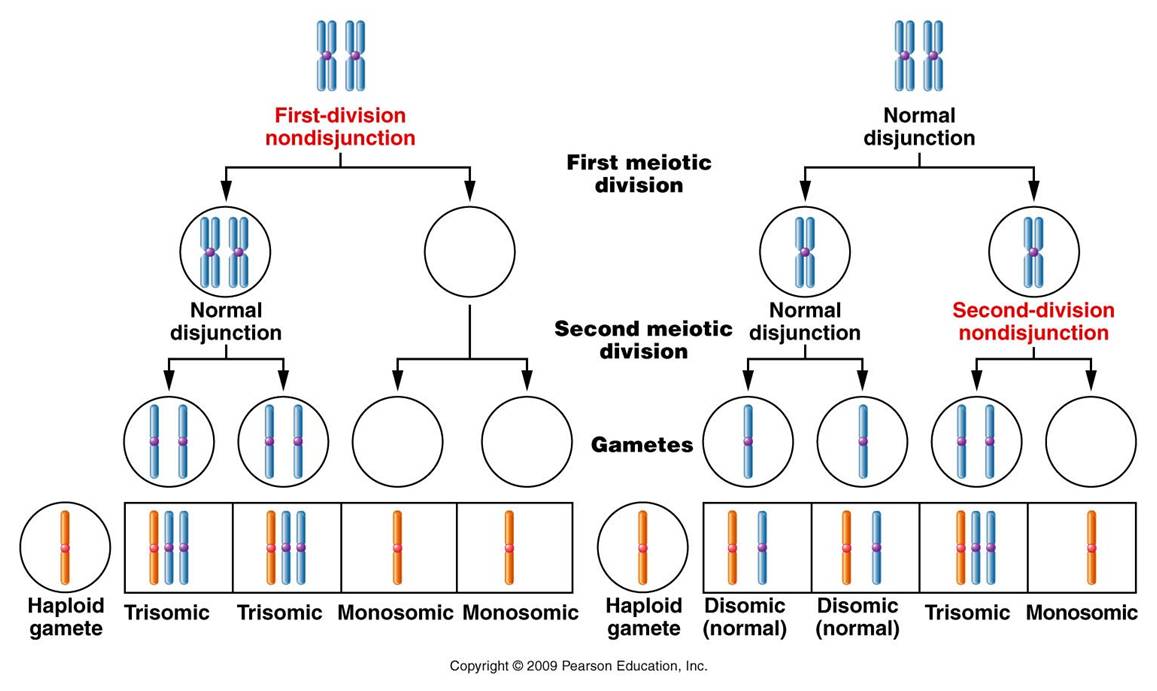
refers to the loss or addition of a single chromosome, not whole sets of chromosomes.
This mutation is called aneuploidy. 'Ploidy' refers
to set, 'eu' means true, and 'an' means not. So,
you have "not a true set".
This
refers to the loss or addition of a single chromosome, not whole sets of chromosomes.
This mutation is called aneuploidy. 'Ploidy' refers
to set, 'eu' means true, and 'an' means not. So,
you have "not a true set".
1. Mechanism:
Non-disjunction - During meiosis, a pair of homologous chromosomes (or sister chromatids) fail to segregate; both go to one daughter cell. So, we end up with a gamete with an extra chromosome, and a gamete that is one chromosome short.
When fertilization occurs with a normal haploid gamete, we end up with a zygote with an extra chromosome - a third chromosome in one homologous 'pair'. (Remember, one gamete inherited BOTH homologs, and then the other parent correctly contributes a single chromosome for this set.) So, this situation is called trisomy (three bodies.) If the other aberrant gamete is involved in a fertilization event, we end up with a zygote with only one chromosome in a given homologous set - this is a monosomy.
2. Human Conditions:
 In humans, these
imbalances are almost always fatal to the developing embryo - they are usually
spontaneously aborted during development (>90% of spontaneous abortions have
chromosomal anomalies). However, some embryos with these conditions can
complete development to birth, and some individuals can survive for decades.
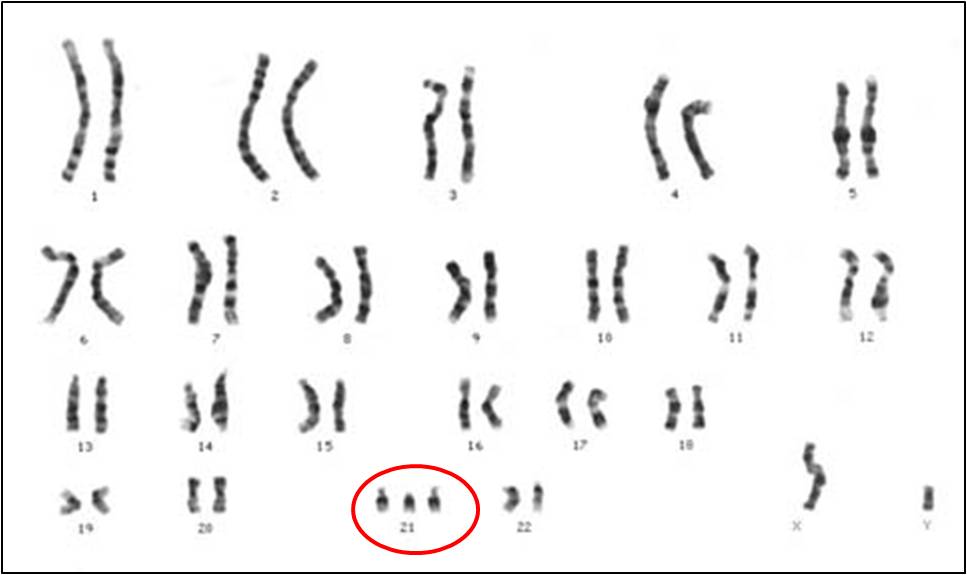
When non-disjunction occurs in the #21 chromosome, TRISOMY 21 occurs. This is
also known as Down's Syndrome. Other autosomal trisomies can survive to birth,
but that is rare and the effects are much more severe. Trisomy 13, Patau Syndrome,
can result in cyclopian eye development (among a host of other deadly anomalies).
Edward's Syndrome, trisomy 18, also causes a rash of typically lethal deformities.
Trisomies in the sex chromosomes are tolerated more often, resulting in XXX,
XXY, and XYY offspring. It is important to appreciate that Down's and sex trisomies
are the trisomies with the LEAST severe effects, where the individual survives
birth and lives for decades. Trisomies should, by chance, be just as likely
for other homologous sets, too; but apparently these trisomies are so devastating
to development that they result in spontaneous abortion.
In humans, these
imbalances are almost always fatal to the developing embryo - they are usually
spontaneously aborted during development (>90% of spontaneous abortions have
chromosomal anomalies). However, some embryos with these conditions can
complete development to birth, and some individuals can survive for decades.
When non-disjunction occurs in the #21 chromosome, TRISOMY 21 occurs. This is
also known as Down's Syndrome. Other autosomal trisomies can survive to birth,
but that is rare and the effects are much more severe. Trisomy 13, Patau Syndrome,
can result in cyclopian eye development (among a host of other deadly anomalies).
Edward's Syndrome, trisomy 18, also causes a rash of typically lethal deformities.
Trisomies in the sex chromosomes are tolerated more often, resulting in XXX,
XXY, and XYY offspring. It is important to appreciate that Down's and sex trisomies
are the trisomies with the LEAST severe effects, where the individual survives
birth and lives for decades. Trisomies should, by chance, be just as likely
for other homologous sets, too; but apparently these trisomies are so devastating
to development that they result in spontaneous abortion.
In humans, the only human monosomy that does, periodically, survive to birth and beyond is the monosomy for the sex chromosome, with 1 X chromosome. This condition, 45, XO, is called Turner's syndrome. No other monosomies are tolerated in humans; all are spontaneously arborted.
1. Mechanism #1: Unequal Crossing-Over:
a. process:
Sometimes during Prophase I of meiosis, the homologs do not line up evenly. If a cross-over event occurs as marked, then the pieces of DNA that are exchanged are not equivalent.
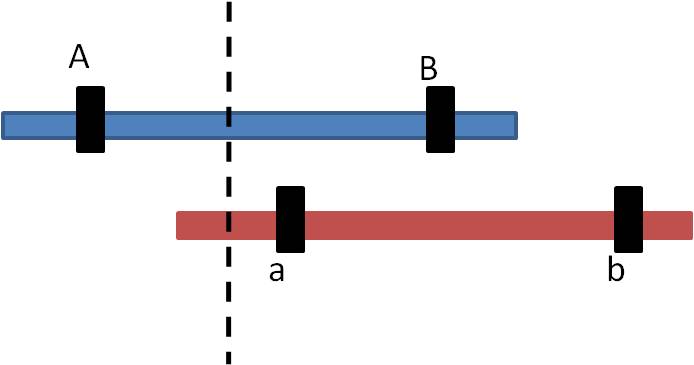
So, the products look like this:
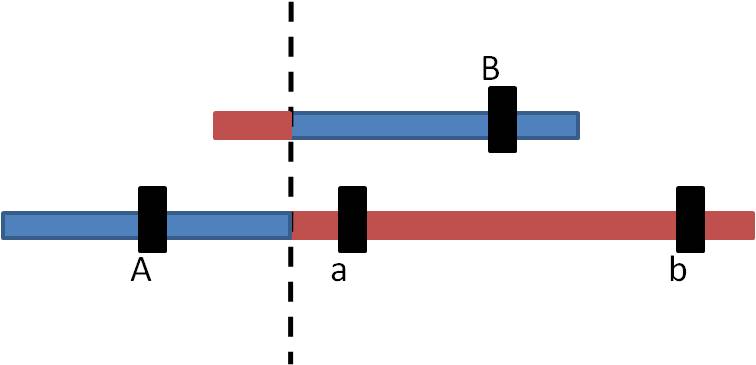
- The "A" locus has been DUPLICATED on the bottom chromosome, and DELETED from the top chromosome.
b. effects:
- Can be bad: DELETION of genes is usually bad... the gene probably did something useful, and now it is gone. At the least, organisms that inherit a deletion MUST express the gene at that lacus that they received from the other parent. So, deletions result in the increased frequency of expression of deleterious recessives inherited from the other parent. That's bad, too. (This is similar to why males expressed X-linked recessives more frequently than females - because they only get one X chromosome, so whatever is on it gets expressed. In that case, the pattern is true for ALL genes on the X chromosome. Here, with a deletion (that can occur on any chromosome), the effect is limited to the genes on the deletion region. But, whatever the organism inherits from the other parent must be expressed.)
DUPLICATION can also be bad. By having three active genes in an other wise diploid genome, the concentration of proteins produced by the duplicated gene may now be "out of whack" (a technical term) with the concentrations of other proteins. Essentially, it is similar to the effects of a trisomy, but at a smaller genetic scale.
- Can be good: Duplication of a gene can be good for two reasons. First, maybe more is better. As we mentioned before, there are many genes for r-RNA production, and many genes for melanin production. How did we get so many genes that are all the same, and code for the same thing? It would be VERY unlikely for random mutation, occurring in two different places in the genome, to create the same sequence of nitrogenous bases just by chance. So, how did it happen? Well, you just saw how - unequal crossing over. And if 'more is better', then organisms with more copies of a gene will be selected for, and these genomes will increase in frequency in the population.
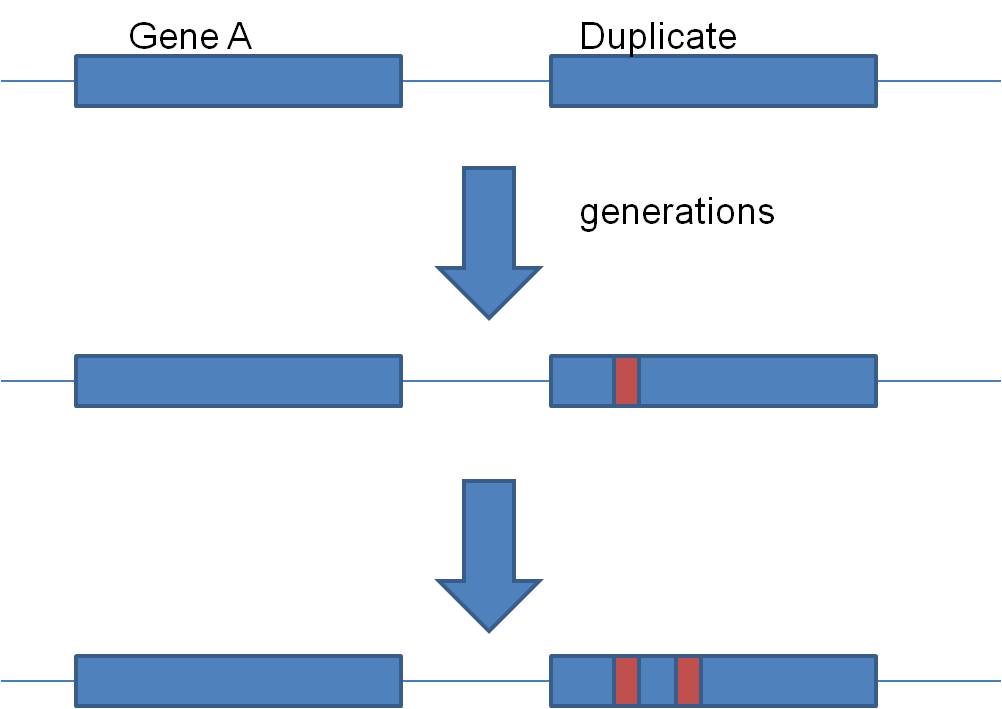 Second, gene duplication is an important source of EVOLUTIONARY NOVELTY - new,
useful variation. You see, it is easier to get something NEW and FUNCTIONAL
by modifying something that already works, rather than building it from scratch.
So, by duplicating a functional gene, evolution can now introduce mutations
into that "copy" and make something new. By tweaking that gene, we might
get something new that also works, but in a slightly different way. This would
produce a "family" of genes that all produce the same type of protein, but proteins
that work a bit differently. We call these families "gene families", and
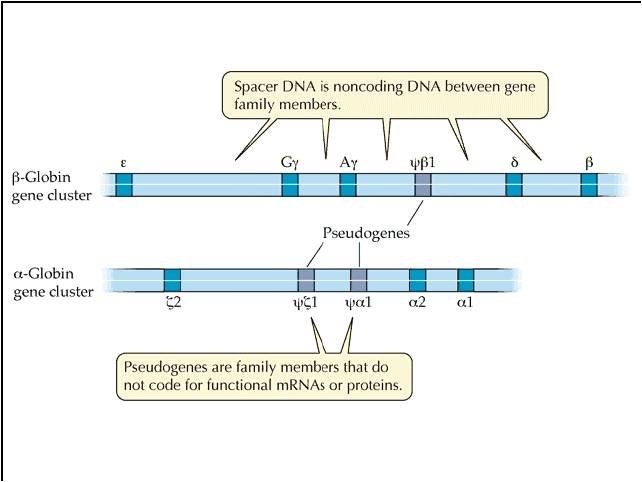
they are quite common. A classic example is the "globin family" of genes.
This includes the alpha, beta, gamma, and myo- globin genes. They are
all similar enough to suggest common ancestry from one ancestral gene, yet different
enough to do slightly different things. And they are all similar enough
in nucleotide sequence that their origin by duplication and subsequent slight
modification is strongly indicated.
Second, gene duplication is an important source of EVOLUTIONARY NOVELTY - new,
useful variation. You see, it is easier to get something NEW and FUNCTIONAL
by modifying something that already works, rather than building it from scratch.
So, by duplicating a functional gene, evolution can now introduce mutations
into that "copy" and make something new. By tweaking that gene, we might
get something new that also works, but in a slightly different way. This would
produce a "family" of genes that all produce the same type of protein, but proteins
that work a bit differently. We call these families "gene families", and
they are quite common. A classic example is the "globin family" of genes.
This includes the alpha, beta, gamma, and myo- globin genes. They are
all similar enough to suggest common ancestry from one ancestral gene, yet different
enough to do slightly different things. And they are all similar enough
in nucleotide sequence that their origin by duplication and subsequent slight
modification is strongly indicated.
There is ANOTHER reason why duplication is an important source of evolutionary novelty, and it relates to an "intelligent design" criticism of evolution. Proponents of intelligent design and creationism suggest that evolution by gradual change is impossible if the intermediate steps are non-functional. For instance, how could the beta-globin gene "evolve from" an alpha globin gene by a gradual process if the intermediate steps are non functional? You see, here's the problem:
 Suppose
alpha globin differs from beta globin by 5 amino acids (I have no idea how many
- this is just an example). In order for the beta globin gene to "evolve",
5 different mutations in the DNA would have to occur, in order to code for five
new amino acids. Now, it is VERY unlikely that all five of those particular
mutations would occur at the same time - mutations are very rare. BUT
(argue the creationists), that's exactly what would have to happen for beta
globin to evolve from alpha globin. Otherwise, they argue, ff we have
one mutation at a time, the alpha globin is turned off and we have NO alpha
globin and NO beta globin - a non-functional intermediate that should be selected
against.
Suppose
alpha globin differs from beta globin by 5 amino acids (I have no idea how many
- this is just an example). In order for the beta globin gene to "evolve",
5 different mutations in the DNA would have to occur, in order to code for five
new amino acids. Now, it is VERY unlikely that all five of those particular
mutations would occur at the same time - mutations are very rare. BUT
(argue the creationists), that's exactly what would have to happen for beta
globin to evolve from alpha globin. Otherwise, they argue, ff we have
one mutation at a time, the alpha globin is turned off and we have NO alpha
globin and NO beta globin - a non-functional intermediate that should be selected
against.
BUT, duplication solves this apparent dilemma. If the alpha gene duplicates, there are now TWO alpha globin genes. Suppose the first mutation occurs in one, and shuts that gene off. So what? The other gene is still right there, cranking out the functinal alpha globin protein. So, the organism with the non-functional duplication is NOT selected against. Rather, they pass their chromosome on to offspring, with the functional and non-functional genes. Generations later, the 2nd, 3rd, 4th, and 5th mutations can accumulate. When the 5th happens, the product is functional beta globin.... and now the organisms that have inherited this mutation have both functional alpha and functional beta globin. Gene duplication is a VERY important mechanism of introducing evolutionarily SIGNIFICANT variation, because it maintains the original gene and its function, while allowing mutations to "play" with the other gene. Mutations that turn that genes off are not selected against, but mutations that improve the gene/protein can still have a positive effect and be selected for. In fact, many regions of non-coding DNA in our genome are "pseudogenes".... regions of non-functional DNA that bear a remarkable similarity in nucleotide sequence to a functional gene. The hypothesis is that they represent a duplicated gene that has since been turned off by mutation.
There is also another bit of evidence
here. We often see members of gene families in the same region of a chromosome
- just as we would expect from unequal crossing-over that places a duplicate
next to its homologous gene. So, both the sequence similiarity and the spatial
proximity that we often see in gene families is strong evidence that unequal
cross-over and gene duplication have been important sources of new useful genes.
 2.
Mechanism #2: Inversions - changing gene order
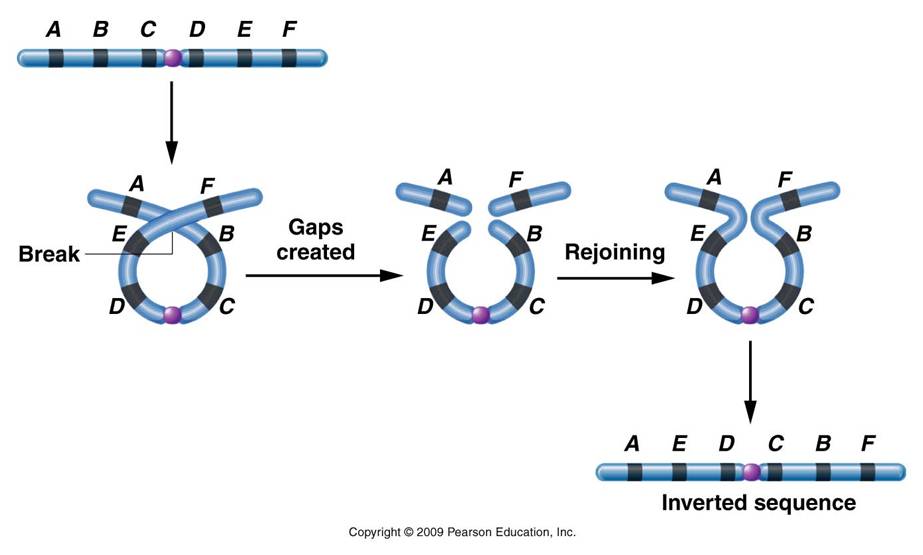
2.
Mechanism #2: Inversions - changing gene orderChromosomes can coil up and cross-over with themselves; inverting a region of genes. The most significant effect that this has on heredity is that subsequent crossing over events between this inversion chromosome and its correctly-ordered homolog now result in non-functional products. The chromosomes that result from crossing over have deletions and duplications. So, the only functional gametes that this organism makes are the parental types - those preserving the arrangement of alleles on each chromosome - including the new arrangment in the inversion region. So, inversions 'stabilize' a set of alleles, preventing their recombination through crossing over. As such, groups of alleles are inherited as a unit, and can be selected for or against as a group. This process can stabilize beneficial combinations of alleles.
 3.
Mechanism #3 - Translocation: moving genes to different chromosomes
3.
Mechanism #3 - Translocation: moving genes to different chromosomes
this is an exchange (or unidirectional transfer) between NON-HOMOLOGOUS chromosomes.
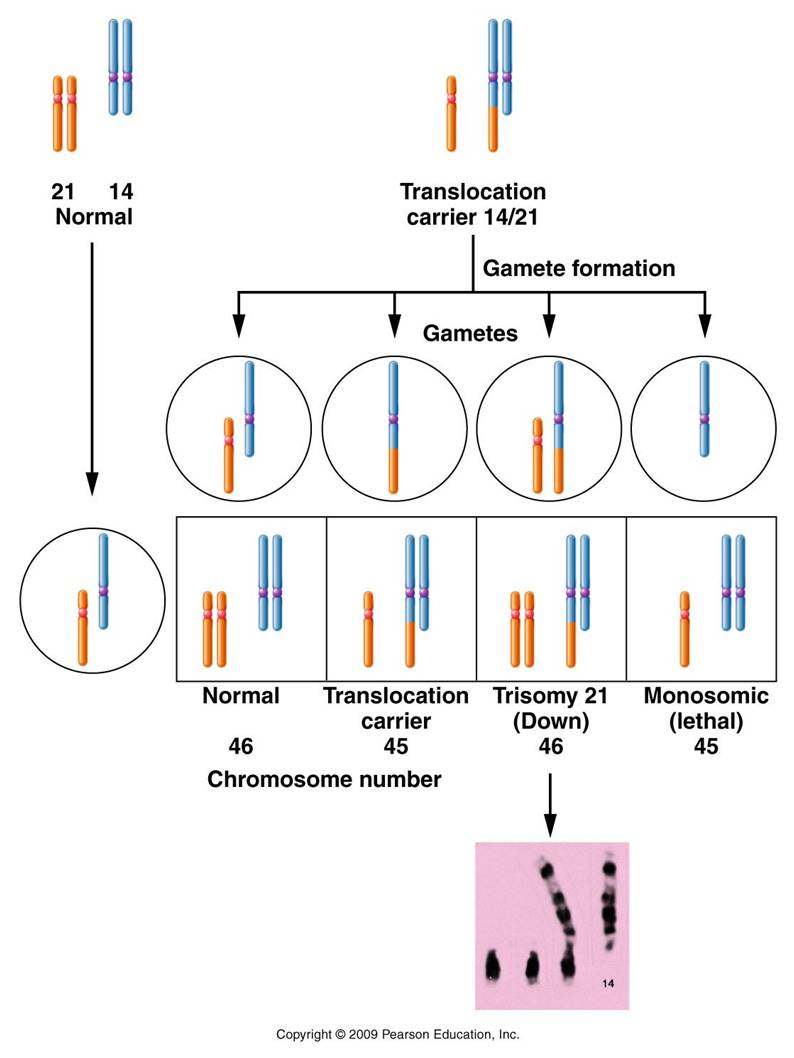
An example is Translocation Down's syndrome. This is where one of the #21 chromosomes gets 'stuck' (translocates) to a #14 chromosome. The individual with this genotype is unaffected - they have the correct DNA content, it's just in a new spatial configuration. But when this individual produces gametes, problems can occur. So... the person has a normal 14, a normal 21, and the translocation chromosome (14+21). The 14's will pair and separate during Meiosis I. If the free 21 goes with the normal 14, you get a normal gamete. But the free 21 could also go with the 14+21 chromosome - resulting in a gamete with two 21's, and another cell with only a 14 (zygote aborted). Fertilization with a normal gamete will produce a zygote with 2 #21's, a free 14, and the 14+21. Two doses of 14 (correct), but 3 doses of 21 (causing Down's syndrome). Trisomy 21 is not heritable. This means that trisomy 21 does not run in families; it is caused by a random non-disjunction error. Translocation Down's, however, IS heritable. So, think about it this way. Suppose you have a sibling with Down's syndrome. If their Down's is caused by trisomy 21, then you would have no greater chance than anyone else in the population of having a child with Down's syndrome. However, if your sibling has translocation Down's, then there is a 50% chance that you - even though you are normal phenotypically - are a carrier of the translocation chromosome. As such, there is a 1/3 chance that you could have a child with translocation Down's.
1. Mechanism #1: Crossing Over within eukaryotic genes can produce new combinations of exons, and new alleles:
- In eukaryotes (and some prokaryotes) with the exon-intron structure of their genes, crossing over WITHIN A GENE, in non-coding intron regions, can create new combinations of exons. This may be a particularly productive way of creating new functional alleles. Why? Well, consider two alleles, A and a, that both function. For simplicity, suppose they are composed of 3 exons each:
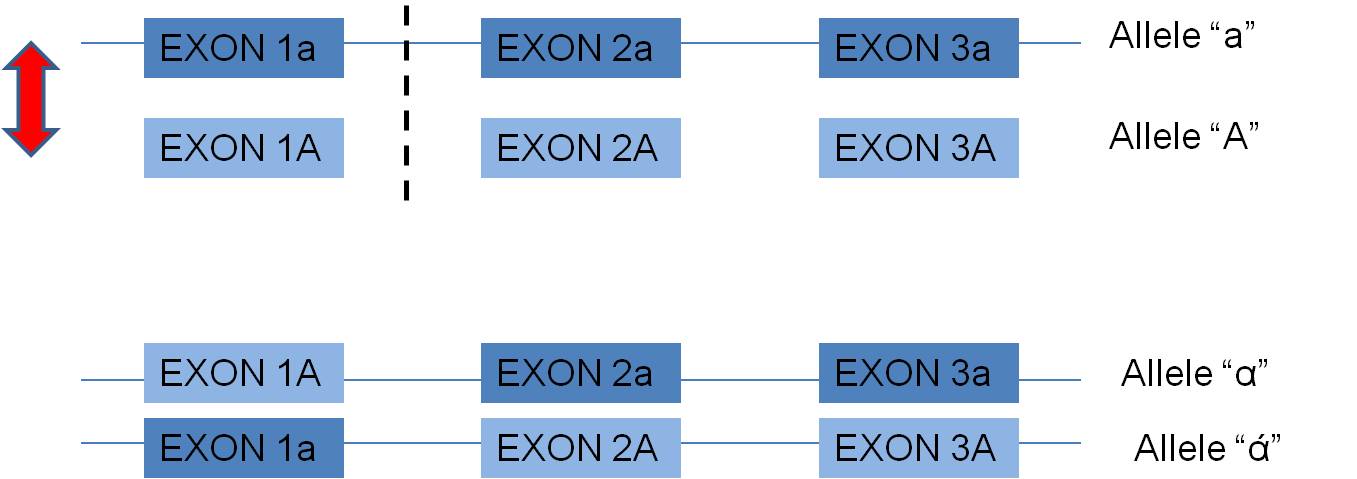
Now, the exons already function with one another. A cross-over between the first and second exon may produce new combinations of exons that also have a high probability of functioning, but maybe in a slightly new way. In fact, many genes across the genome seem to share particular exons that encode for a particular functional domain of proteins. It is far more likely that these are homologous similiaries arising from "exon shuffling" than the independent but parallel creating of these identical sequences by mutation.
2. Mechanism #2: Point Mutations - changes in nucleotide sequence
a. Addition/deletion:
When DNA is being replicated, the DNA polymerase may erroneously "skip" a base,
or erroneously add an extra base into the newly forming strand. If this
is the sense strand that will later be transcribed, this will cause the addition
or deletion of a base in the m-RNA. This change will "shift" the 3-base
reading frame during translation, changing every codon downstream: 
So, from the point of the addition or deletion, every amino acid that follow will change. So, these "frameshift" mutations usually have a deleterious effect because they change a whole bunch of amino acids.
b. Substitutions:
- during DNA replication, an improper base can be substituted while the new strand is being synthesized. Many of these errors are corrected during 'DNA proof-reading' and repair that occurs in G1 and G2, but some (1 in a million) slip by. This change in the DNA (if in an exon) would change one base in the m-RNA, and change one m-RNA codon. This could change an amino acid - but if you look at the genetic code you will see that most AA's are encoded by more than one three base codon. So, for instance, CUU, CUA, CUG, and CUC all code for the amino acid "leucine". So, a change in the third position of the DNA sequence encoding these codons would have NO EFFECT on the amino acid:
Original DNA = GAT ....... original
RNA = CUA ....... amino acid = leucine
mutant DNA = GAA ........ mutant RNA = CUU ...... amino
acid is STILL LEUCINE
So, some mutations are "silent", even if they occur within an exon of a gene.
However, other mutations can change
the amino acid:
Original DNA = GAT ....... original RNA = CUA ....... amino acid = leucine
mutant DNA = GTT ........ mutant RNA = CAA ...... amino
acid is GLYCINE

The smallest change that is possible in DNA is a single nucleotide, and this may cause the smallest change possible in a protein - a change in a single amino acid. These small changes may be deleterious, neutral, or beneficial. And, AS YOU KNOW, the value of a gene may depend on the environment.
Consider the sickle cell anemia example that we discussed before. Hemoglobin consists of two alpha globin molecules and two beta globin molecules. The allele for the normal beta globin chain contains 146 amino acids, with glutamine as the 6th amino acid. The sickle cell hemoglobin contains the SAME alpha chains as normal hemoglobin, but a mutant form of the beta chain. In the mutant form, the 6th amino acid is valine. This difference in one amino acid is caused by a change in one DNA nucleotide. As we have discussed, it is beneficial in the tropics but deleterious in the temperate zone.
We began the unit with Darwin's model
of evolution:
Sources of variation
Agents of change
unknown
Natural selection.
As a consequence of our modern understanding of heredity and genetics, we have learned quite a bit about variation AND evolution. Our model, a this point in the class is:
Sources of variation
Agents of Change
MUTATION:
-New Genes:
Natural Selection
point mutation
Mutation (polyploidy can make new species)
exon shuffling
RECOMBINATION:
- New Genes:
crossing over
-New Genotypes:
-crossing over
- independent assortment
Sources of Variation
Agents of Change
MUTATION:
-New Genes:
Natural Selection
point mutation
Mutation (polyploidy can make new species)
RECOMBINATION:
- New Genes:
exon shuffling
-New Genotypes:
-crossing over
- independent assortment
 In
the early 20th century, at the same time that T. H. Morgan was studying mutations
and creating linkage maps, other biologists were considering the evolutionary
implications of this new knowledge regarding genetic variation. They appreciated
that individuals do not evolve - evolution is a process that occurs at the population
level. For example, as a consequence of differential reproductive success among
individuals in a population, the range of phenotypes and their relative frequencies
in the population will change over time. Individuals are born, life, reproduce
(maybe) and die. As a result of passing on their genes at different frequencies,
the genetic structure of the population changes over time (evolution). Two biologists,
G. Hardy and W. Weinberg, constructed a model to explain how the genetic structure
of a population might change over time.
In
the early 20th century, at the same time that T. H. Morgan was studying mutations
and creating linkage maps, other biologists were considering the evolutionary
implications of this new knowledge regarding genetic variation. They appreciated
that individuals do not evolve - evolution is a process that occurs at the population
level. For example, as a consequence of differential reproductive success among
individuals in a population, the range of phenotypes and their relative frequencies
in the population will change over time. Individuals are born, life, reproduce
(maybe) and die. As a result of passing on their genes at different frequencies,
the genetic structure of the population changes over time (evolution). Two biologists,
G. Hardy and W. Weinberg, constructed a model to explain how the genetic structure
of a population might change over time.
Their model begins by constructing an 'equilibrium' model - a model of what the genetic structure would look like, and how it would behave, if there was NO CHANGE over time. (We can liken this to a "statistical null hypothesis of no effect"). Then, an actual population is compared to this model, to see whether the population is evolving or not.
Our first step is to describe the genetic structure of a population; we need to do this before we can model what it would do over time. The genetic structure of a population is defined by the gene array and the genotypic array. To understand what these are, some definitions are necessary:
1. Definitions:
- Evolution: a change in the genetic structure of a population
- Population: a group of interbreeding organisms that share
a common gene pool; spatiotemporally and genetically defined
- Gene Pool: sum total of alleles held by individuals in a population
- Genetic structure: Gene array and Genotypic array
- Gene/Allele Frequency: % of alleles at a locus of a particular
type
- Gene Array: % of all alleles at a locus: must sum to 1.
- Genotypic Frequency: % of individuals with a particular genotype
- Genotypic Array: % of all genotypes for loci considered;
must = 1.
2. Basic Computations - Determining the Genotypic and Gene Arrays:
 The
easiest way to understand what these definitions represent is to work a problem
showing how they are computed.
The
easiest way to understand what these definitions represent is to work a problem
showing how they are computed.
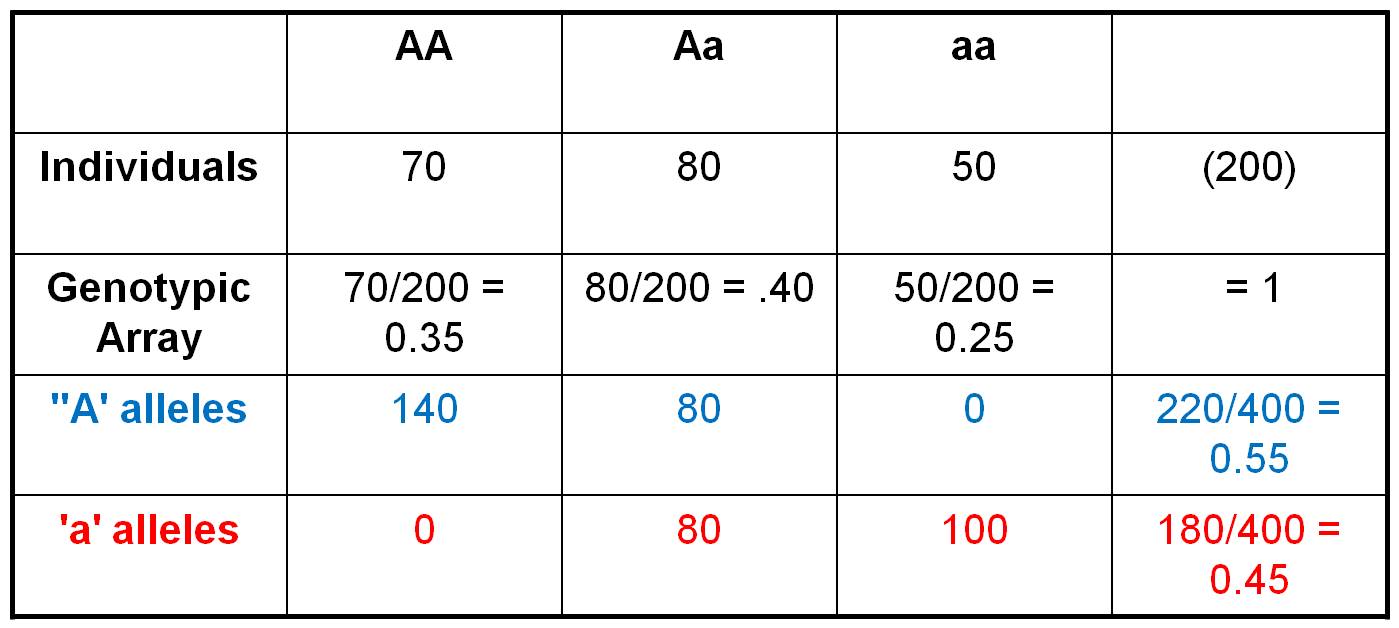
Consider the population shown to the right, in which there are 70 AA individuals, 80 heterozygotes, and 50 aa individuals. We can easily calculate the Genotypic Frequencies by dividing each of these values by the total number of individuals in the population. So, the Genotypic Frequency of AA = 70/200 = 0.35. If we account for all individuals in the population (and haven't made any careless math errors), then the three genotypic frequencies should sum to 1.0. The Genotypic Array would list all three genotypic frequencies: f(AA) = 0.35, f(Aa) = 0.40, f(aa) = 0.25. A Gene Frequency is the % of all genes in a population of a given type. This can be calculated two ways. First, let's do it the most obvious and direct way, by counting the alleles carried by each individual. So, there are 70 AA individuals. Each carries 2 'A' alleles, so collectively they are 'carrying' 140 'A' alleles. The 80 heterozygotes are each carrying 1 'A' allele. And of course, the 'aa' individuals aren't carrying any 'A' alleles. So, in total, there are 220 'A' alleles in the population. With 200 diploid individuals, there are a total of 400 alleles at this locus. So, the gene frequency of the 'A' gene = f(A) = 220/400 = 0.55. We can calculate the frequency of the 'a' alleles the same way. The 50 'aa' individuals are carrying 2 'a' alleles each, for a total of 100 'a' alleles. The 80 heterozygotes are each carrying an 'a' allele, and the 140 AA homozygotes aren't carrying any 'a' alleles. So, in total, there are 180 'a' alleles out of a total of 400, for a gene frequency f(a) = 180/400 = 0.45. The gene array presents all the gene frequencies, as: f(A) = 0.55, f(a) = 0.45.
There is a faster way to calculate the gene frequencies in a population than adding up the genes contributed by each genotype. Rather, you can use these handy formulae:
f(A) = f(AA) + f(Aa)/2
f(a) = f(aa) + f(Aa)/2
So, to calculate the frequency of a gene in a population, you add the frequency of homozygotes for that allele with 1/2 the frequency of heterozygotes. In our example, this would be:
f(A) = 0.35 + 0.4/2 = 0.35 + 0.2 = 0.55
f(a) = 0.25 + 0.4/2 = 0.25 + 0.2 = 0.45
Wow... that's a lot faster.
1. Goal:
The goal of the "Hardy-Weinberg Equilibrium Model" (HWE) is to describe what the genetic structure of the population would be if NO evolutionary change occurs. Working independently, Hardy and Weinberg realized that the gene frequencies in a population will NOT change - will remain in EQUILIBRIUM - if the following conditions are met:
- there is random mating
- no selection
- no mutation
- no migration
- and the population is infinitely large.
And, they realized that a population will reach an equilibrium in GENOTYPIC frequencies, too, after one generation of meeting these expectations. And, for as long as these conditions are met, a population will NOT EVOLVE. Let's see how they came by these conditions.
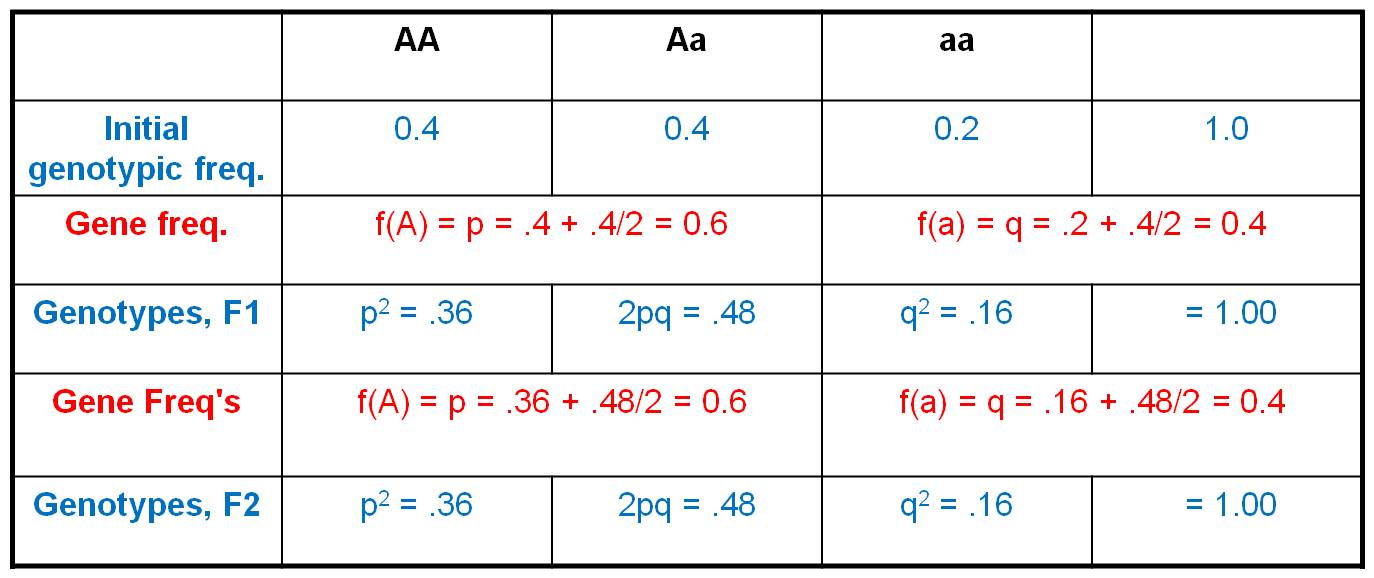 2.
Example:
2.
Example:
Consider an initial population, with a genotypic array as shown. The gene frequencies are:
A = 0.4 + (0.4/2) = 0.6
a = 0.2 + (0.4/2) = 0.4
Now, consider this gene pool in which 60% of the alleles are 'A' and 40% of the alleles are 'a' (as defined by the gene frequencies). The gene frequencies represent the frequencies of gametes carrying these gens; so 60% of sperm are 'A', 40% are 'a', and likewsie for eggs.
So, now we employ the HWE model. IF the population mates at random, then we can use the product rule to determine the probability of any two gametes coming together. The propability that and 'A' sperm fertilizes an 'A' egg = 0.6 x 0.6 = 0.36. And of course, this is the only way to produce an 'AA' zygote. The frequency of 'AA' zygotes (the F1 offspring) produced by this population should be 0.36. Likewise, the probability that an 'a' sperm fertilizes an 'a' egg = 0.4 x 0.4 = 0.16. And again, this is the only way to make an 'aa' zygote, so the total frequency of 'aa' zygotes in the F1 will be 0.16. Now, there are two ways to make an 'Aa' zygote: an 'A' sperm can fertilize an 'a' egg (probability = 0.6 x 0.4 = 0.24), and an 'a' sperm can fertilize an 'A' egg (also with a probability of 0.4 x 0.6 = 0.24). So, the total frequency of Aa zygotes in the F1 will be 2 x 0.24 = 0.48. If we generalize, and let f(A) = p and f(a) = q, then the genotypic frequencies under HWE can be calculated as: f(AA) = p2, f(Aa) = 2pq, and f(aa) = q2.
What is the genetic structure of the population in the F1? Well, f(A) = f(AA) + f(Aa)/2 = 0.36 + 0.48/2 = 0.36 + 0.24 = 0.6. And, f(a) = f(aa) + f(Aa)/2 = 0.16 + 0.48/2 = 0.4. So, the gene frequencies did not change. And, if these organisms produce gametes at these gene frequencies and mating is random, then F2 zygotes should be formed at the frequencies of f(AA) = 0.36, f(Aa) = 0.48, and f(aa) = 0.16. Look familiar? Indeed, after one generation of random mating, the population has reached an EQUILIBRIUM - constant gene and genotypic frequencies over time.
Now, of course, these calculations will only be true IF the population mates at random. AND, they will only be true if there is no mutation. If 'A' alleles are mutating into 'a' alleles, then the gene frequencies will not be 0.6 and 0.4, and calculations based on these numbers will not be correct. So, we must assume NO MUTATION. Likewise, we can't have any migration; we can't have 1000 AA individuals migrate into our population, or that would change the gene frequencies, too; and our predictions based on frequencies of 0.6 and 0.4 would be incorrect. So, we must assume NO MIGRATION, too.
So, at this point we have zygotes at the frequencies shown in the "Genotypes, F1" row. In order for there to be no change in the genetic structure of the population, there must be NO SELECTION. In other words, all genotypes must have the same probability of survival and reproduction. Only then will they contribute gametes at frequencies of p = 0.6 and q = 0.4. (If there were selection, and if AA individuals were the only zygotes to survive to reproduce, for instance, then the gene frequencies would change and our predictions based on frequencies of 0.6 and 0.4 would not be correct).
And finally, this model will only be explicitly true for populations that are infinitely large: because that is the only time when we can be garaunteed that predictions based on random chance will be exactly met. (Think about it this way... suppose I give you a coin that is absolutely perfectly balanced. It IS PERFECTLY BALANCED. And suppose I ask you, "how many times do you have to flip that coin to be ABSOLUTELY SURE of producing a 50:50 ratio of heads to tails? Well, if you only flip it four times, you know that, just by chance, you would often get 3 heads and a tail or 3 tails and a head. And even if you flip it 10,000 times, you might get 5001 heads and 4999 tails, even though the coin is perfectly balanced. To be absolutely garaunteed that the predictions of this probabilisitic model will be met exactly, you must flip the coin an infinite number of times. Obviously, this is a theoretical constraint because no population is infinitely large. But this is a theoretical model of no change, so we can employ theoretical expectations. The same is true of our 'expectation' of a perfectly balanced coin - this expectation will only be met, for sure, in an infinitely large sample. Yet we continually employ that expectation for a perfectly balanced coin, even in finite samples. So, if you flip the coin 20 times, how many heads would you expect? Your answer of 10 is a theoretical expectation.
So, that is why these assumptions exist. It is only when ALL these are met that the genetic structure of a population will not change. It is only when ALL these assumptions are met that a population will NOT evolve. Wow. That should seem rather amazing. It is only when these assumptions are ALL met that a population WON'T change. If any of these assumptions is not met, a population's genetic structure WILL change... and that is evolution. So, from this analysis, we should expect populations to evolve - it is only under a rare combination of events (no, mutation, no selection, no migration, random mating, and an infinitiely large population) that evolution WON'T happen.
3. Utility
- If no real populations can explicitly meet these assumptions, how can the model be useful? For instance, no real population is infinitely large, so how can the model be useful? We use it for COMPARISON. This model describes what the genotypic frequencies should be IF the population was in equilibrium. If the real genotypic frequencies are not close to these expectations, then the population is not in HWE.... it is evolving. And if a population is not in HWE, then the population must be violating one of the assumptions of the HWE model. Think about that. The HWE is only 'true' if all the assumptions are being met. If your real population differs from the model, then one of the assumptions must not apply to your real population. This narrows your focus on WHY the real populations isn't behaving randomly... and it might identify WHY the population is evolving.... which is a biologically interesting question.
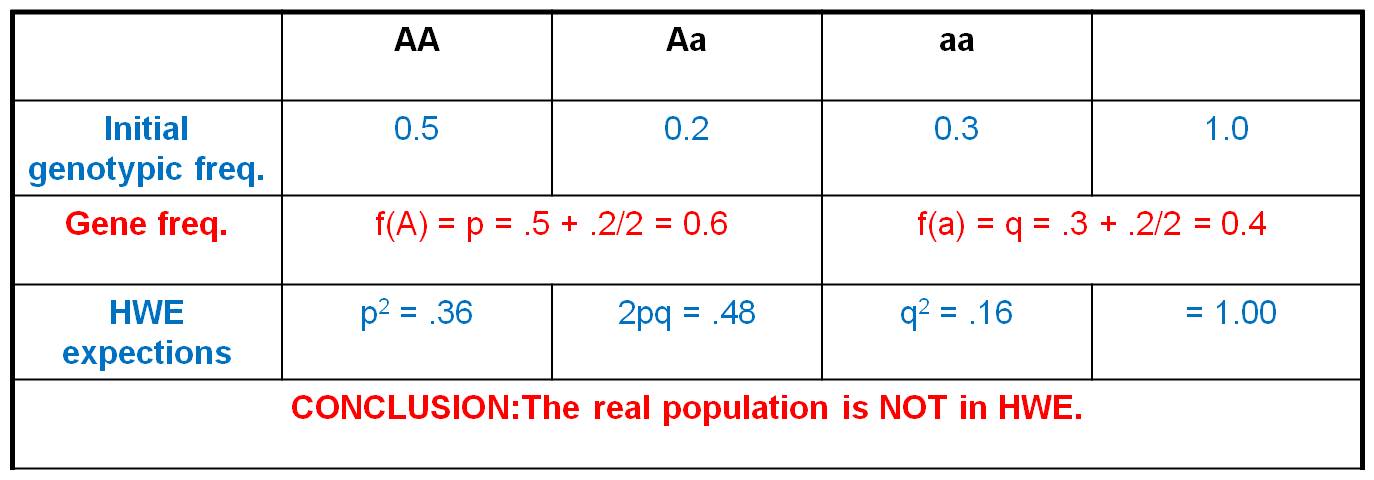 -
Again, the coin analogy applies. No REAL coin is probably exactly perfectly
balanced. But, if I give you a coin and ask you how balanced it is, you flip
it a few times and compare its behavior to WHAT YOU WOULD EXPECT FROM A PERFECT
COIN (50:50 RATIO). Even though a perfectly balanced coin may not exist, we
can use this theoretical model as a benchmark, to compare the behavior of real
coins. Many real coins act in a manner that is consistent enough with
the expectations from a perfectly balanced coin that we are willing to use them
AS IF they were perfectly balanced. The Hardy Weinberg Equilibrium
Model is the same... it is a theoretical model of no change against which we
can measure real populations.
-
Again, the coin analogy applies. No REAL coin is probably exactly perfectly
balanced. But, if I give you a coin and ask you how balanced it is, you flip
it a few times and compare its behavior to WHAT YOU WOULD EXPECT FROM A PERFECT
COIN (50:50 RATIO). Even though a perfectly balanced coin may not exist, we
can use this theoretical model as a benchmark, to compare the behavior of real
coins. Many real coins act in a manner that is consistent enough with
the expectations from a perfectly balanced coin that we are willing to use them
AS IF they were perfectly balanced. The Hardy Weinberg Equilibrium
Model is the same... it is a theoretical model of no change against which we
can measure real populations.
If HWE can be assumed, then the frequency of recessive diseases can be assumed to equal q2, and the frequency of carriers in the population can be estimated like this:
1) The frequency of hemachromatosis worldwide is 1/450. If we assume that hemochromatosis is caused by a recessive gene (q), and if we assume the population is in HWE with respect to this trait, then q2 = 1/450 = 0.002. So, we take the square-root of both sides to find q = 0.047. Well, if q = 0.047, and if p + q = 1, then p = 1 - 0.047 = 0.953.
2) If q = 0.047 and p = 0.953, then the frequency of heterozygous carriers = 2pq = 0.09. So, we estimate that 9% of the population are carriers.
Now, you might say, "but we just determined that HWE would be unusual; so why would we assume it is true for a given gene?" Well, a deleterious gene has already been largely weeded out of a population, so selection against the few alleles that are left is really weak. Indeed, this condition may not influence reproductive success, anyway (NO SELECTION). In addition, we don't select mates based on whether they have hemochromatosis (I bet you NEVER asked your date if they have hemochromatosis, for example!!), so we can assume there is RANDOM MATING in the population with respect to this trait. And although the human population is not infinite, it is really big (~7 BILLION), so the effect of sampling error is probably very small. Mutation is very rare, so the effects of mutation are likely to be very small. And if we are making an estimate based on the whole human population, then there can be no 'migrants' coming in from somewhere else (Martians?). So, in some cases, we can reasonably assume a population might be in HWE for a given gene. Of course, we could be wrong... and we would test that prediction by sampling individuals in the population and determining the frequency of heterozygotes genetically. But at least we would have a working hypothesis.
1. How can a new species form by polyploidy, and why is this more common in plants than animals?
2. Alpha and beta globin molecules are VERY similar to one another in nitrogenous base sequence. What does this suggest about where they came from? How might this have happened?
3. Describe two reasons why duplications may be advantageous.
4. How are new genes produced? List two ways (in eukaryotes).
5. How is translocation Down's caused?
6. Why are substitution mutations likely to have a less pronounced effect tha addition or deletion mutations?
7. Outline the causes of genetic variation.
8. What are the five assumptions of the Hardy-Weinberg Equilibrium Model?
9. Consider the following population:
| AA | Aa | aa | |
|---|---|---|---|
| Number of Individuals | 60 |
20 |
20 |
10. If the HWE model does not describe any real population, how can it be useful?